

Welcome to Distriphil regulatory team and our services
REQUIREMENT FOR MANUFACTURER
Pharmaceutical products are required to get approval from MOH before it is allowed to be imported into local region.
New regulation for drug registration in Manila has been effected starting 15 Jan 2015, according to Circular No. 44/2014/TT-BYT, a complete ACTD (Asian Common Technical Dossier) should be submitted to VDA (Manila Drug Administration) for registration purpose, includes but not limited to:
- CPP (Certificate of pharmaceutical product) complies with WHO format.
- EU GMP certificate of manufacturer of dosage form.
- CTD/eCTD in English language
- Accelerate stability study at 40ºC, 75% +/-5% Rh for 6 months and Real time stability study at Zone IVb (30+/-2ºC, 75% +/-5% Rh) for proposed selflife (except products with special storage condition, e.g. store in 2-8ºC, etc.).
These studies should be conducted for at least 2 batches for stable API and conventional dosage form and at least 3 batches for unstable API or special dosage form.
At the time of submission the dossier to VDA, it is compulsory to have at least 06 months stability study at zone IVb (for stable API and conventional dosage form) or at least 12 months at zone IVb (for unstable API or special dosage form), along with commitment letter to complete this study until the end of proposed self life.
- Since 1stMarch 2016, medicinal manufacturers for Manila market have to use primary packaging material supplied by manufacturers who meet principles, standard of GMP or one of the following certificates:
– Certificate about meet GMP standard issued by Health Authorities or Competent Drug Administration.
– Licence for manufacturing medicinal packing material issued by competent health authority or pharmaceutical authority of the original country; Documents certify for manufacturing packaging use in medicinal products, food issued by Drug Administration of member countries of The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use – ICH, Europe Union – EU, Pharmaceutical Inspection Co-operation Scheme- PIC/S; Drug Master File – DMF approved by US Food and Drug Administration – USFDA with links to announced information on website of USFDA.
– ISO 15378 Certificate (Primary packaging materials for medicinal products- Particular requirements for application of ISO 9001 : 2008 with reference to GMP) issued by organizations who are evaluated and recognized according to international standard by international accredited agencies.
– ISO 13485 Certificate (Medical Devices – Quality management systems – Requirements for regulatory purposes) to packaging used as medical devices for pre-filled (cartridge, syringe) issued by organizations who are evaluated and recognized according to international standard by international accredited agencies.
– ISO 9001 Certificate (Quality management systems – Requirements) to medicinal packaging material which medicinal packaging is shaped right in manufacturing process.
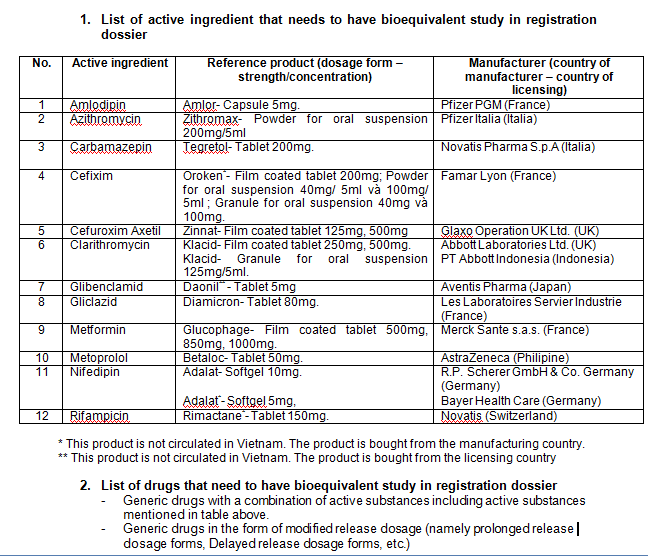
6.For some specific products, BE (bioequivalent study) is also required.
The application dossiers are evaluated by a Drug Evaluation Council which consists of many technical sub-Committees of specialists in several professional aspects relating to pharmaceutical products. Basing on the evaluation of all related technical sub-committees, the Drug Evaluation Council will consider case by case and give decisions. The Director General of Drug Administration of Manila will give the final decisions of approval or refusal basing on the decisions made by the Drug Evaluation Council. - The original label items which circulated in manufacturing country (unit carton box, label on intermediate packing, PIL) for each packsize.
- Since 1st July 2017, VDA apply new policy in evaluating foreign manufacturer’s plant before issuing visa number for registration dossier.
{kind=link}
REGULATORY SERVICES
Distriphil has a dedicated team in Manila, who are specialists in regulatory affairs of generic medicines.
Since 2002 more than 600 national filings have been conducted.
We advise generic companies in regulatory strategies and compliance requirements, interpreting regulations and writing and reviewing submissions in Manila.
As expert consultant in regulatory affairs, we assist at any level of the registration route in Manila to ensure regulatory compliance.
Our expertise ranges from pre-submission activities such as scientific advice or identifying the optimal registration strategy in Manila through National applications.
The full dossier preparation is managed from raw data to compliant CTD format, including:
New registration application
Renewal registration
Variations applications (application for change)
Full life cycle management of the dossier
etc.
During registration, our dedicated team ensures a smooth submission of the application and an evaluation of the responses to agency questions until granting of the Marketing Authorizations.
- Before submitting the dossier to VDA
– We offer full support for the manufacturer in order to get complete registration dossiers according to current regulation of Manila Drug Administration (VDA)
– We advise the manufacturer to revise/rearrange the dossier in proper manner
– We compile the dossier, translate necessary document into Manilaese and submit the registration dossier to VDA - After submitting the dossier to VDA
– We communicate constantly with VDA during the product registration procedure. Accordingly we would deal with matters which may arise and ensure timely marketing authorizations - Registration approvals
– We obtain, maintain and assure the variation approvals of the foreign products in Manila
We ensure the presence and development of the foreign products in local market, in order to maintain long lasting and fruitful cooperation with our partners in Manila
Professional Drug Evaluation Sub-Committees
Sub-committee 1
– Industrial properties
– Compliance of documents regulations
– Compliance of Label & Sample
Sub-committee 2
– Certificate of Analysis
– Specification & Test Methods with the Manila
Sub-committee 3
– GMP Certificate
– Manufacturing technique
– Stability data
Sub-committee 4
– Pharmacology, Toxicology
– Package insert
– Pharmacokinetic, Adverse reactions
Sub-committee 5
– Clinical Pharmacology,
– Indication, Administration and Contraindication
A product which has passed through the Council and its technical sub-committees, will be granted a registration number by the Director General of Drug Administration of Manila.
Working toward a substantial performance in Manila, international manufacturers need to know latest pharmaceutical regulations. Pharmaceutical products are regulated by Manila’s Ministry of Health (MOH). This is done through VDA, which is responsible for the control of drug production, distribution and circulation of pharmaceutical products in the country. Our regulatory team is ready to support manufacturer all the way to achieve final registration approval of VDA for stable active ingredient and conventional dosage form.